Become a member and receive career-enhancing benefits
Our top priority is providing value to members. Your Member Services team is here to ensure you maximize your ACS member benefits, participate in College activities, and engage with your ACS colleagues. It's all here.
Become a member and receive career-enhancing benefits
Our top priority is providing value to members. Your Member Services team is here to ensure you maximize your ACS member benefits, participate in College activities, and engage with your ACS colleagues. It's all here.
Late Onset Ornithine Transcarbamylase Deficiency Triggered by Total Parenteral Nutrition Use in the Surgical ICU
January 31, 2024
15 MinPrintShare
Bookmark
Abstract
Background
Ornithine transcarbamylase deficiency is a rare X-linked error of metabolism affecting the urea cycle. Male neonates classically develop hyperammonemia due to decreased breakdown of carbamoyl phosphate. Females may not present with typical symptoms due to incomplete enzyme function causing partial ornithine transcarbamylase deficiency (pOTCD). These patients may have mild or subclinical symptoms until a catabolic stressor precipitates fulminant disease.
Summary
A 66-year-old female underwent an emergent ileocolectomy with end ileostomy to treat a small bowel obstruction due to ileal stricture, a complication of Crohn's disease. Her postoperative course was complicated by dehydration due to high output ileostomy. She also had parastomal pyoderma gangrenosum complicated by a necrotizing soft tissue infection of the abdominal wall with an enterocutaneous fistula. Total parenteral nutrition (TPN) was started, and a rapid decline in mental status necessitated intubation. In seeking etiology for acute encephalopathy, results included hyperammonemia and normal liver enzyme levels. TPN was discontinued, and the patient's condition improved. Brain MRI demonstrated changes consistent with hyperammonemia-related encephalopathy, suggesting pOTCD. This case presents a notable precipitation of pOTCD due to TPN administration. The patient had no significant history besides an aversion to meat, exhibiting no prior symptoms of hyperammonemia. Genetic testing was considered but ultimately unnecessary in the acute setting as it would not have changed clinical decision-making. This rare but life-threatening complication of TPN requires immediate recognition to reverse the process. The diagnosis can be made clinically with cessation of TPN.
Conclusion
This case describes a patient who experienced an acute mental status change with isolated hyperammonemia instigated by total parenteral nutrition administration in the surgical intensive care unit (ICU). Isolated hyperammonemia in the context of normal liver enzymes should raise suspicion of occult metabolic disorders in patients with acute mental status change after TPN administration. Genetic testing may not always be indicated in the acute care setting to guide medical management.
The patient was a 66-year-old woman who presented to the emergency department with a complaint of an abdominal wall wound. Two months prior to presentation, she underwent an emergent end ileostomy and ileocecectomy for Crohn's disease, indicated by am ileal stricture causing a small bowel obstruction. For the preceding three weeks, she was managed as an outpatient for dehydration due to high output from her ileostomy and a peristomal lesion consistent with pyoderma gangrenosum. Her medical history was otherwise significant for hyperlipidemia, hypothyroidism, and avoidance of meat due to malaise. This avoidance of meat was not included in her initial history. Surgical history included previous incisions and drainage of perirectal abscesses and a colonoscopy demonstrating slight narrowing at the terminal ileum.
Figure 1. Peristomal Pyoderma Gangrenosum Documented Three Weeks Post-operation, Significant for Ulceration, Purulence, and Necrosis. Published with Permission
Photo taken prior to development of abdominal wall necrotizing soft tissue infection.
At her current presentation, symptoms included three days of vomiting, increased purulence, foul smell, and ulceration of the peristomal wound. There was diffuse abdominal wall pain, greatest intensity localized to the right-sided ulceration (Figure 1). A CT scan of the abdomen revealed an intraabdominal fluid collection. Broad-spectrum IV antibiotics were started, and the patient was transferred urgently to the operating room for excisional debridement of the abdominal wall necrotizing soft tissue infection. A computed tomography-guided abdominal drain was placed to drain the intraabdominal abscess, with 310 mL of purulent fluid aspirated. Drain output transitioned to greenish-yellow fluid consistent with succus, and an enteral fistula was diagnosed.
The patient was malnourished and had inadequate nutrition intake since her initial operation. TPN was initiated postoperative day 2, including 100 g of protein daily. Within 24 hours, the patient complained of vague feelings of malaise, progressing to generalized weakness. She had an acute mental status change, was responsive to only painful stimuli, and had a new rightward gaze deviation. She also had hypertension, tachycardia, and tachypnea. The patient was intubated and mechanically ventilated for impending respiratory failure. Immediate EKG, chest radiography, and labs were significant for compensated metabolic acidosis and isolated hyperammonemia to 303 (normal range 11–51 µMOL/L), with otherwise normal liver function tests. CTA head demonstrated no acute bleeding and normal perfusion. An EEG demonstrated encephalopathy due to status epilepticus for which anticonvulsant medication was started. Oral and rectal lactulose were administered with a transient improvement of altered mental status followed by further decline. The ammonia level continued to climb, peaking at 472 µMOL/L. Interval head CT demonstrated no evolving pathology.
TPN was discontinued. Ammonia levels normalized to less than 50 µMOL/L. In the following two weeks, the patient underwent continued treatment for intraabdominal sepsis and status epilepticus. On postoperative day 12, an MRI of the head showed bilateral diffuse restricted diffusion changes consistent with metabolic encephalopathy, seen in hyperammonemia. Her ICU course was complicated by failed extubation, deep venous thrombosis, and mucous plugging. The patient was eventually discharged from the hospital on postoperative day 35. At the time of discharge, she was tolerating gastrostomy feeding, a tracheostomy collar without mechanical ventilation, and had improved cognition. She was instructed to continue her antiepileptic medication at a skilled nursing facility and follow up with colorectal and neurology services as an outpatient.
The patient was ultimately diagnosed with pOTCD. Genetic testing and urine orotic acid levels were not pursued in the management of her hospital care, as a result, would not have altered clinical decision-making and would have led to unnecessary costs.
Discussion
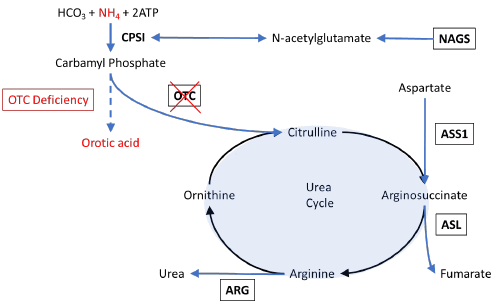
Ornithine transcarbamylase deficiency (OTCD) is an X-linked inborn error of metabolism affecting the urea cycle. The deficiency occurs as a mutation on the Xp21.1 gene, manifesting as decreased enzyme expression in hepatocytes and the gastrointestinal tract.1 It is the most common urea cycle disorder affecting 1/17,000 people in the United States.2 Ornithine transcarbamylase functions to convert carbamyl phosphate and ornithine into citrulline (Figure 2). When ornithine transcarbamylase is deficient, ammonia accumulates due to decreased breakdown of carbamyl phosphate. This has been shown to disrupt aquaporin synthesis and increase the production of astrocyte glutamine which can cause brain edema and encephalopathy.3 Conversion of ornithine into citrulline is prevented, which results in the accumulation of orotic acid.
Figure 2. pOTCD and Urea Cycle. Published with Permission
Description of the urea acid cycle with elements involved in ornithine transcarbamylase deficiency is outlined in red. OTC: ornithine transcarbamylase; CPSI: carbamoyl phosphate synthetase I; ATP: adenosine triphosphate; NAGS: N-acetylglutamate synthase; ASS1: argininosuccinate synthetase I; ASL: argininosuccinate lyase; ARG: arginase
Classical symptoms present as hyperammonemia in male neonates, causing lethargy, poor appetite, and coma.4 Affected females may harbor atypical symptoms due to selective X inactivation and residual enzyme function, known as partial ornithine transcarbamylase deficiency (pOTCD).5 Patients who are heterozygous develop milder symptoms, often having a history of self-limiting protein intake.6 They may develop acute hyperammonemic episodes during major illness, surgery, pregnancy, corticosteroid and valproic acid administration, prolonged fasting, increased protein intake, and total parenteral nutrition (TPN).6
Patients with pOTCD can present with acute hyperammonemia and normal liver function when enduring catabolic stress.6 Major illness, surgery, pregnancy, corticosteroid and valproic acid administration, prolonged fasting, increased protein intake, and TPN are known triggers of pOTCD.6 In our patient, the absence of abnormal liver enzymes was key in evaluating more esoteric causes of hyperammonemia.
Meat avoidance due to malaise is also characteristic in patients with pOTCD.6 A key question in this patient's workup was deciding whether to order urine orotic acid or genetic testing. In the context of her necrotizing soft tissue infection and acute hospital care, confirming pOTCD would not change clinical outcomes or our management. She underwent the appropriate testing for acute encephalopathy in a post-surgical patient, and TPN was appropriately discontinued. Her improvement in neurologic status provided empiric evidence that the underlying cause had been addressed.
Upon discharge, the patient was offered genetic testing to confirm the presence of pOTCD. She declined due to advanced age, lack of biological children, and high cost. Alternative forms of testing include protein load and allopurinol challenge, but these carry high risks of metabolic decompensation, false positives, and false negatives.7 DNA-based analysis with sequencing and Southern blot analysis of the gene may only detect 70% of relevant mutations.8 Eighty-six percent of mutations causing pOTCD are single base substitutions which are often novel to each family line, further hindering the capabilities of genetic testing.8 There is ongoing research to determine biomarkers for ureagenesis, elemental isotope ratios, and proton magnetic resonance spectroscopy for unidentifiable cases by DNA testing.7,9
Increased protein intake has been described as a specific inciter of previously unexplained hyperammonemic encephalopathy in patients with pOTCD.10 In one case series, the two most important factors in treatment were the early identification of hyperammonemia as the cause of metabolic encephalopathy and the initiation of hemodialysis.10 This case suggests that if the offending agent is identified early in the clinical course, hemodialysis may not be necessary. However, the use of lactulose without a hepatic cause of encephalopathy was unwarranted when superior agents such as sodium benzoate and sodium phenylacetate could have been used.
Hyperammonemia associated with pOTCD can manifest in a spectrum of neurological outcomes, including encephalopathy, subclinical or partial seizures, and status epilepticus.3,11,12 These manifestations are caused by metabolic disturbances as well as structural changes to the brain. This can be visualized on brain MRI with hypomyelination, gliosis of the grey matter, and cystic changes of the white matter.12
Conclusion
This is a case of acute encephalopathy due to underlying pOTCD precipitated by TPN. We recommend caution with TPN formulation for patients who avoid high-protein foods. Include pOTCD in the differential diagnosis for encephalopathy and hyperammonemia absent liver dysfunction following initiation of TPN.
Lessons Learned
Catabolic stress or a typical protein load from TPN can precipitate complications of pOTCD. Protein intolerance or meat avoidance is pertinent history to obtain prior to starting TPN. Urea cycle enzyme deficiencies should be considered in cases of acute encephalopathy with concurrent TPN administration.
Authors
Shahmanyan Da; Katz Ga,b; Nowak Ea,c; Bower KLa,b
Author Affiliations
Virginia Tech Carilion School of Medicine and Research Institute, Roanoke, VA 24016
Carilion Clinic Department of Surgery, Roanoke, VA 24014
Carilion Clinic Department of Medicine, Roanoke, VA 24014
Corresponding Author
Katie L. Bower, MD, MSc, FACS 1906 Belleview Avenue SE Ste. 322 Roanoke, VA 24014 Email: klbower1@carilionclinic.org
Disclosure Statement
The authors have no conflicts of interest to disclose.
Funding/Support
The authors have no relevant financial relationships or in-kind support to disclose.
Received: April 12, 2021 Accepted: August 5, 2021
References
Brassier A, Gobin S, Arnoux JB, et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: a series of 90 patients. Orphanet J Rare Dis. 2015;10:58. Published 2015 May 10. doi:10.1186/s13023-015-0266-1
Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127-170.
Machado MC, Pinheiro da Silva F. Hyperammonemia due to urea cycle disorders: a potentially fatal condition in the intensive care setting. J Intensive Care. 2014;2(1):22. Published 2014 Mar 13. doi:10.1186/2052-0492-2-22
Levin B, Abraham JM, Oberholzer VG, Burgess EA. Hyperammonaemia: a deficiency of liver ornithine transcarbamylase. Occurrence in mother and child. Arch Dis Child. 1969;44(234):152-161. doi:10.1136/adc.44.234.152
Scaglia F, Zheng Q, O'Brien WE, et al. An integrated approach to the diagnosis and prospective management of partial ornithine transcarbamylase deficiency. Pediatrics. 2002;109(1):150-152. doi:10.1542/peds.109.1.150
Gropman AL, Fricke ST, Seltzer RR, et al. 1H MRS identifies symptomatic and asymptomatic subjects with partial ornithine transcarbamylase deficiency. Mol Genet Metab. 2008;95(1-2):21-30. doi:10.1016/j.ymgme.2008.06.003
Barkovich E, Gropman AL. Late Onset Ornithine Transcarbamylase Deficiency Triggered by an Acute Increase in Protein Intake: A Review of 10 Cases Reported in the Literature. Case Rep Genet. 2020;2020:7024735. Published 2020 Apr 25. doi:10.1155/2020/7024735
Bogdanovic MD, Kidd D, Briddon A, Duncan JS, Land JM. Late onset heterozygous ornithine transcarbamylase deficiency mimicking complex partial status epilepticus. J Neurol Neurosurg Psychiatry. 2000;69(6):813-815. doi:10.1136/jnnp.69.6.813
Wang FS, Goh DLM, Ong HT. Urea cycle disorder presenting as bilateral mesial temporal sclerosis - an unusual cause of seizures: a case report and review of the literature. J Med Case Rep. 2018;12(1):208. Published 2018 Jul 15. doi:10.1186/s13256-018-1750-8